Cystic Fibrosis
- Non-communicable
- Genetic disorders
- Autosomal recessive

Cystic fibrosis (CF) is an aggressive life-limiting autosomal recessive disease with a high rate of mortality. Mutation in the cystic fibrosis conductance regulator (CFTR) gene is the sole origin of CF. CFTR gene encodes a protein that functions cAMP-activated chloride (Cl-) channel. Mutation in CFTR gene leads to less or no production of CFTR protein that results in the defective or reduced transport of Cl- and bicarbonate (HCO3-) ions in multiple organs. Organs affected due to mutation include gut, lungs, liver, pancreas and reproductive organs, however, the lungs are mostly affected organs. CF lung disease is characterized by the accumulation of viscous mucus, chronic inflammation, and persistent bacterial infections in the lungs. The thick, viscous mucus leads to clogging in the respiratory linings resulting in stuffy nose, wheezing and breathlessness. Persistent infection-directed inflammation leads to permanent structural damages of the respiratory linings that eventually results in respiratory failure and death. CF is highly prevalent among the Caucasian population from developed countries including UK, Europe, North America and Australasia. Until 2005, the life expectancy of patients with CF was around 28 years, however, with the advancement of diagnosis and treatment, the median survival time of patients with CF increased to 37 years over the past decade.
What is cystic fibrosis and why surfing helps. This video was provided courtesy of the Mauli Ola Foundation. Learn more at http://www.mauliola.org/.
Clinical Features
In the past, a diagnosis of CF was based primarily on clinical signs and symptoms. However, with the introduction of CF newborn screening (NBS) in many countries including the United States, a majority of newly diagnosed cases are minimally symptomatic or asymptomatic infants. Screening involves procedures such as immunoreactive trypsinogen (IRT) biomarker testing on dried blood spots as well as testing for one or more CFTR gene mutations. In many cases, the diagnosis can be confirmed by a high chloride concentration in a follow up sweat test.
Given the variation in disease severity and age of onset, CF diagnosis can be a challenge. Patients with CF can present with manifestations such as respiratory symptoms, chronic sinusitis, pancreatitis and male infertility. Moreover, patients with CF lung disease present with persistent cough with viscous mucus, wheezing, repeated airway infection, leading to bronchiectasis, gas trapping, hypoxaemia, and hypercarbia.
Pathogenesis
CF is a monogenic disease caused by mutation in CFTR gene. Patients with CF suffer from intense systemic inflammation characterized by increased serum acute phase proteins, high antibody titres to respiratory
pathogens
Pathogens
A biological agent such as viruses, bacteria or fungi that cause a disease.
, atopy and heightened Th2 responses. Initially, it was thought that CFTR was largely expressed by the airway epithelial cells (AECs), however, expression and function of CFTR had been reported to other inflammatory cell types such as monocytes, macrophages, T cells, mast cells. Molecular studies suggest a negative feedback loop between functional CFTR and inflammation. However, it is still unknown why CF inflammation never stops.
Airway epithelial cells (AECs)
The airway epithelium acts as structural and immunological barriers to the inhaled
pathogens
Pathogens
A biological agent such as viruses, bacteria or fungi that cause a disease.
. Airway surface liquid (ASL) is a low viscous mucus layer containing a number of proteins including mucin and anti-microbial peptides that tightly regulate antimicrobial activity, mucociliary transport and trapping and clearance of the inhaled
pathogens
Pathogens
A biological agent such as viruses, bacteria or fungi that cause a disease.
. Mutation in CFTR gene not only distort the structural integrity of the airway but also abolish Cl- ion transport leading to the production of viscous mucus which trap the respiratory
pathogens
Pathogens
A biological agent such as viruses, bacteria or fungi that cause a disease.
resulting in chronic inflammation in the respiratory linings. Airway epithelial cells (AECs) of the CF lungs release enormous amount of pro-inflammatory cytokines those chemoattract neutrophils and macrophages.
Neutrophils
Neutrophils play crucial role in CF disease
pathogenesis
Pathogenesis
The biological mechanism that leads to a state of disease. It can refer to the origin and development of a disease, as well as whether it is recurrent, chronic and acute.
. Higher neutrophil counts had been observed in the bronchoalveolar (BAL) fluid and CF specimens of CF patients. Microbial killing ability of neutrophils had been compromised with non-functional CFTR. Elevated neutrophil elastase activity in CF airway is thought not only to be the underlying reason for structural damage of airway linings but also compromise activation of adaptive immune cells.
Monocytes/Macrophages
Monocytes maintain a circulatory pool of immune effector cells. During infection, they migrate to the tissues and differentiate into either macrophages or dendritic cells to mount innate and adaptive immune responses respectively. Macrophages with their pro-inflammatory (M1) and anti-inflammatory (M2) subsets maintain balance between inception and resolution of respiratory inflammation. Truncated form of CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication.
had been observed in both CF monocytes and macrophages. Monocytes from CF patients demonstrate enhanced inflammatory
cytokine
Cytokine
A broad category of small proteins released by a range of cells (including immune cells like macrophages, B lymphocytes, T lymphocytes) that act through receptors and are important for a range of processes including cell signaling, inhibition, maturation and proliferation.
profile with defective phagocytic ability. Macrophages in CF had been found highly pro-inflammatory without their anti-inflammatory attributes.
Comorbidities
Repeated and chronic infection with bacteria, particularly with Staphylococcus aureus and Pseudomonas aeruginosa are common in CF. Episodes of infection increase with age and are associated with neutrophilic inflammation leading to lung damage, bronchiectasis that leads to
fibrosis
Fibrosis
Excessive tissue forming to compansate during organ repair. This overcompansation can often serve to obstruct the organ from functioning properly.
during adulthood.
Cellular Characteristics
CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication.
is expressed at the apical membrane of almost all cell types, including epithelial cells, monocytes, neutrophils, mast cells. Following synthesis, CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication.
enters into the endoplasmic reticulum (ER) and golgi apparatus for checking the proper folding and glycosylation of the
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication.
. The fully matured and functional CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication.
is then transported to the
plasma
Plasma
Blood plasma is the yellow-colored portion of the blood carrying cells and proteins throughout the body.
membrane to acts as Cl- ion channel. Mutation in CFTR gene leads to less or no production, trafficking and function of CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication.
. Cellular effects due to the mutation vary with cell types. For instance, mutation in CFTR leads to aberrant Cl- ion channel activity in airway epithelial cells, defective bacterial clearance by monocytes and macrophages, excessive inflammatory mediator release by neutrophils and mast cells.
Excessive neutrophil counts along with reduced antimicrobial activity and dysregulated
cytokine
Cytokine
A broad category of small proteins released by a range of cells (including immune cells like macrophages, B lymphocytes, T lymphocytes) that act through receptors and are important for a range of processes including cell signaling, inhibition, maturation and proliferation.
production by neutrophils are the hallmark features of CF lung inflammation. An inverse relationship was observed between dysfunctional CFTR protein and cellular redox state, ER stress and mucin overproduction by AECs. Enhanced release of CCL20 (
macrophage
Macrophage
A variety of white blood cell which serves the primary function of digesting cellular debris.
recruitment chemokine) and higher
macrophage
Macrophage
A variety of white blood cell which serves the primary function of digesting cellular debris.
count had been observed in CF airways. Reduced acidification of lysosomes in macrophages leads to defective clearance of bacteria from the respiratory linings. Failure of bacterial clearance in the lungs results in chronic infections that later lead to remodelling and thickening of airway lining and eventually failure of lung functions.
Genetics
Types of mutations in CFTR
The mutation in CFTR gene was first reported in 1989. From then, more than 2000 mutations have so far been reported in human CFTR gene which are broadly categorized into six different classes according to their effects on the maturation and function of CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication.
(Figure 1). The most common mutation is the F508del (also known as ΔF508) mutation, with prevalence varying depending on the geographical region.
- Class I mutations result in no production of CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication. . - Class II mutations, the most common mutations among patients, are associated with impaired post-translational modification, such as, misfolding, inadequate glycosylation leading to the defective trafficking of CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication. to the cell surface. - Despite the presence of full-length CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication. to the cell surface, class III mutations alter channel gating and result in reduced Cl- channel activity. - Class IV mutations lead to the regular production of CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication. with reduced Cl- permeability. - Class V mutations lead to the production of minimal copies of functional CFTR.
- Class VI mutations negatively affect the
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication. stability causing an accelerated turnover of CFTR protein Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication. at cell surfaces thereby reduce the quantity.
Recently, 11 new rare CFTR mutations have been reported among patients from the Indian subcontinent that increased the spectrum of CFTR mutations worldwide.
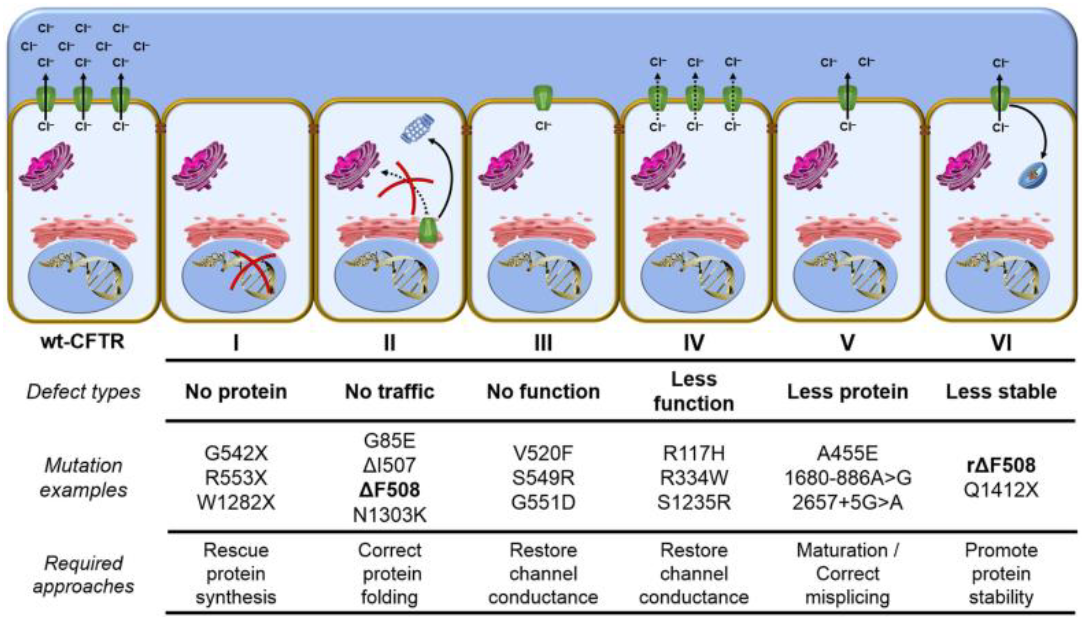
Figure 1: Classes of CFTR mutations. Adopted from Lopes-Pacheco et al.
Treatment
Conventional treatments
The conventional treatment options for CF patients aim to slow lung disease and its progression, to improve airway clearance. Since chronic infection in the lungs and exaggerated respiratory inflammation are the hallmark features of CF, antibiotics and anti-inflammatory drugs are frequently chosen for CF patients. Antibiotics are mainly given to the CF patients to eradicate the bacterial infections. Tobramycin is highly recommended for CF patients in US to delay colonization of bacteria in the lungs. Significant improvement of lung functions, decreased bacterial and neutrophil counts, reduced pulmonary inflammation and weight gain had been observed following tobramycin administration. In addition, tobramycin had been shown to promote translational read-through of the premature stop codon (class I CFTR mutation) and generate the full-length functional CFTR
protein
Protein
Large biomolecules, which consist of one or more long chains of amino acid residues. They perform a vast array of functions within organisms including transporting molecules, providing cellular structure and catalysising processes such as DNA replication.
in AECs from CF patients. However, long-term usage of tobramycin increases the risk of renal failure and promote antibiotic resistance.
Azithromycin (AZM) is a macrolide class antibiotic that are known to reduce acute respiratory exacerbations, to improve lung function weight gain and quality of life in CF patients without correcting CFTR mutation. Macrolides are known to show anti-inflammatory actions by reducing neutrophilic inflammation. The antibiotic activity of macrolides resides in the “sugar” moiety attached to the macrolide ring, however removing this may not alter the anti-inflammatory actions. Non-antibiotic macrolides (NAMs) had been shown to restore the phagocytic function of airway macrophages. Therefore, whether NAMs would be beneficial in CF is under consideration. Ibuprofen as an anti-inflammatory drug is recommended for long-term treatment for CF patients. Inhaled dornase-α is recommended for CF patients with moderate to severe CF to reduce the viscosity of sputum. Hypertonic saline and inhaled β-agonists are recommended for CF patients over 6 years of age.
Mutation-specific treatments
Although conventional treatment options showed significant improvement of the lung function, however, they are unable to repair the deleterious effects of the mutations in the CFTR gene. Therefore, mutation-targeted drugs that can restore normal CFTR function are in demand. A number of small molecules known as CFTR corrector and CFTR potentiators had been identified by high throughput screening (HTS). CFTR correctors increase the delivery of functional CFTR to the cell surface (effective for class II mutations), while CFTR potentiators increase channel gating (effective for class III mutations). Ivacaftor has been approved by FDA as CFTR potentiator for the patients carrying class II G551D allele and class IV mutations. Ivacaftor had been shown to enhance channel activity, improve lung function, lowers sweat chloride level and thereby improve the clinical outcomes in patients carrying class III mutation. Lumacaftor is a promising CFTR corrector that has been shown to selectively correct class II mutations with improved CFTR post-translational modification, trafficking to the cell surface and enhanced Cl- ion secretion. The maximal effect has been observed after 24hr of treatment. Unfortunately, monotherapy of lumacaftor in patients carrying homozygous class II mutations failed to show significant improvement of CFTR function in AECs and significant changes in lung functions. Furthermore, patients with heterozygous (class II/III) mutations require treatment with both Ivacaftor and Lumacaftor to achieve clinical benefit. Combinational therapy of Ivacaftor and Lumacaftor showed enhanced channel gating, improved lung function and lowered frequency of hospitalization compared to monotherapy or control group.
Gene therapy
Gene therapy is an attractive tool to deliver the correct copy of CFTR gene via viral vector into the individual cells of the lungs. Since almost all structural and immune cells are affected in CF lung diseases, delivery of the correct copy of CFTR gene remained challenging. Clinical trial of gene therapy did not show substantial improvement of lung function. Adverse effects following gene therapy set this tool as hope more than a reality. CRISPR/Cas9 is a very recent and advanced gene delivery tool. Ongoing research is being conducted for development of novel CRISPR/Cas9-based CFTR gene delivery system.
Epidemiology
CF is one of the most common life threatening recessively inherited disorders in the world. Those most affected are children, typically diagnosed before the age of two. The disease predominantly affects Caucasian populations compared with other ethnic groups. It is difficult to estimate CF prevalence, especially in non-Caucasian populations. Additionally, less severe cases may be diagnosed later in life. In predominantly Caucasian populations, the birth prevalence of CF is around 1 in 2500 live births. These figures may vary, with Scandinavian countries such as Sweden and Norway reporting a much lower rate. Finland has a CF birth prevalence as low as 1 in 40000 births. As of 2015, around 30000 people were living with CF in the United States and over 90% of individuals were Caucasian.
The life expectancy of CF has increased in recent years with advancements in diagnosis and care. The median survival time of patients with CF is around 37 years worldwide. This can vary depending on severity of disease and a key factor is the type of CFTR mutation. The F508del mutation is present in over 60% of CF cases worldwide, although prevalence varies depending on geographical region and ethnicity. Among the broad range of mutations present in CF cases globally, F508del is by far the most common, followed by G451X and G551D mutations.
Additional Resources

The Mauli Ola Foundation
The Mauli Ola Foundation (MOF) is a nonprofit organization dedicated to providing hope and confidence to individuals living with genetic diseases. Harnessing the healing powers of the ocean, they introduce surfing and ocean-based activities as natural therapies. They are honored to share their knowledge of the ocean, family values and community.
http://www.mauliola.org/

Rock CF
Rock CF is a community Thousands strong changing the face of what living with Cystic Fibrosis looks like and giving those living with CF the tools to not only survive, but thrive.
THEIR FOOTPRINT:
• Empowering CF people to live a healthy life.
• Bringing a positive spin to the demanding challenges CF poses though speaking engagements.
• Heightening cystic fibrosis awareness through the Rock CF apparel line and events.
• Outfitted over 300 people through the Kicks Back program.
• Sponsored entry fees for people with CF to participate in running races.
• Inspired lifestyle changes on a small scale that can make big difference.
• Provide resources for advocacy work to families.
• Fund research through the Cystic Fibrosis Foundation, Mayo Clinic and others.
http://letsrockcf.org/
References
- "Foundation, C.F. Patient registry: annual data report, 2012. Bethesda, MD. Cystic Fibrosis Foundation, 2013 (2013)."
- "Bell, S.C. et al. Cystic fibrosis in Australia, 2009: results from a data registry. The Medical journal of Australia 195, 396-400 (2011)."
- "Sorio, C. et al. Defective CFTR expression and function are detectable in blood monocytes: development of a new blood test for cystic fibrosis. PloS one 6, e22212 (2011)."
- "Amaral, M.D. CFTR and chaperones: processing and degradation. J Mol Neurosci 23, 41-48 (2004)."
- "Guerra, L. et al. Antibiotic therapy affects functional behaviour in cystic fibrosis blood mononuclear cells. Eur Respir J (2015)."
- "Saiman, L. Microbiology of early CF lung disease. Paediatr Respir Rev 5 Suppl A, S367-369 (2004)."
- "Armstrong, D.S. et al. Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol 40, 500-510 (2005)."
- "Hubeau, C., Puchelle, E. & Gaillard, D. Distinct pattern of immune cell population in the lung of human fetuses with cystic fibrosis. The Journal of allergy and clinical immunology 108, 524-529 (2001)."
- "Starner, T.D., Barker, C.K., Jia, H.P., Kang, Y. & McCray, P.B., Jr. CCL20 is an inducible product of human airway epithelia with innate immune properties. American journal of respiratory cell and molecular biology 29, 627-633 (2003)."
- "Di, A. et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nature cell biology 8, 933-944 (2006)."
- "Tarique, A.A. et al. CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J Cyst Fibros (2017)."
- "Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front Pharmacol 7, 275 (2016)."
- "Sharma, H., Jollivet Souchet, M., Callebaut, I., Prasad, R. & Becq, F. Function, pharmacological correction and maturation of new Indian CFTR gene mutations. J Cyst Fibros 14, 34-41 (2015)."
- "Bruscia, E.M. & Bonfield, T.L. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. J Innate Immun (2016)."
- "Bruscia, E.M. & Bonfield, T.L. Innate and Adaptive Immunity in Cystic Fibrosis. Clin Chest Med 37, 17-29 (2016)."
- "Martinez, F.O., Gordon, S., Locati, M. & Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177, 7303-7311 (2006)."
- "Tarique, A.A. et al. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. American journal of respiratory cell and molecular biology 53, 676-688 (2015)."
- "Sturges, N.C. et al. Monocytes from children with clinically stable cystic fibrosis show enhanced expression of Toll-like receptor 4. Pediatr Pulmonol 45, 883-889 (2010)."
- "Bruscia, E.M. et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. American journal of respiratory cell and molecular biology 40, 295-304 (2009)."
- "Geller, D.E. Aerosol antibiotics in cystic fibrosis. Respir Care 54, 658-670 (2009)."
- "Flume, P.A. et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. American journal of respiratory and critical care medicine 176, 957-969 (2007)."
- "Prayle, A. & Smyth, A.R. Aminoglycoside use in cystic fibrosis: therapeutic strategies and toxicity. Current opinion in pulmonary medicine 16, 604-610 (2010)."
- "Smyth, A. et al. Case-control study of acute renal failure in patients with cystic fibrosis in the UK. Thorax 63, 532-535 (2008)."
- "Wolter, J. et al. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 57, 212-216 (2002)."
- "Hodge, S. et al. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. American journal of physiology. Lung cellular and molecular physiology 312, L678-L687 (2017)."
- "Van Goor, F. et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences of the United States of America 106, 18825-18830 (2009)."
- "Rowe, S.M. et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. American journal of respiratory and critical care medicine 190, 175-184 (2014)."
- "Van Goor, F. et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences of the United States of America 108, 18843-18848 (2011)."
- "Wainwright, C.E. et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med (2015)."
- "Moss, R.B. et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum Gene Ther 18, 726-732 (2007)."
- "Farrell, P. M., et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. The Journal of pediatrics, 181, S4-S15 (2017)."
- "Ross, L. F. Newborn screening for cystic fibrosis: a lesson in public health disparities. The Journal of pediatrics, 153(3), 308-313 (2008)."
- "Marshall, B. C., et al. Cystic Fibrosis Foundation patient registry: annual data report 2015. Cystic Fibrosis Foundation, Bethesda, MD (2015)."
- "Walters, S. A. & Mehta A.N. \"Epidemiology of cystic fibrosis\". Cystic fibrosis (2007): 21-45."
- "LiPuma, J. J. The changing microbial epidemiology in cystic fibrosis. Clinical microbiology reviews, 23(2), 299-323 (2010)."
- "Mehta, G., et al. Cystic fibrosis across Europe: EuroCareCF analysis of demographic data from 35 countries. Journal of Cystic Fibrosis, 9, S5-S21 (2010)"